 Oligomerization and the formation of aggregates of misfolded proteins are common to many genetic and sporadic forms of neurodegenerative diseases. Although some of these misfolded proteins are caused by mutations directly within disease-related proteins, such as in the polyglutamine expansion diseases and some forms of familial Alzheimer’s disease, the mechanisms underlying protein misfolding in many sporadic forms of neurodegenerative diseases remain unknown. We used a forward genetic approach to identify novel genes that, when their function is disrupted, cause the accumulation of misfolded proteins in neurons prior to their death.
Oligomerization and the formation of aggregates of misfolded proteins are common to many genetic and sporadic forms of neurodegenerative diseases. Although some of these misfolded proteins are caused by mutations directly within disease-related proteins, such as in the polyglutamine expansion diseases and some forms of familial Alzheimer’s disease, the mechanisms underlying protein misfolding in many sporadic forms of neurodegenerative diseases remain unknown. We used a forward genetic approach to identify novel genes that, when their function is disrupted, cause the accumulation of misfolded proteins in neurons prior to their death.
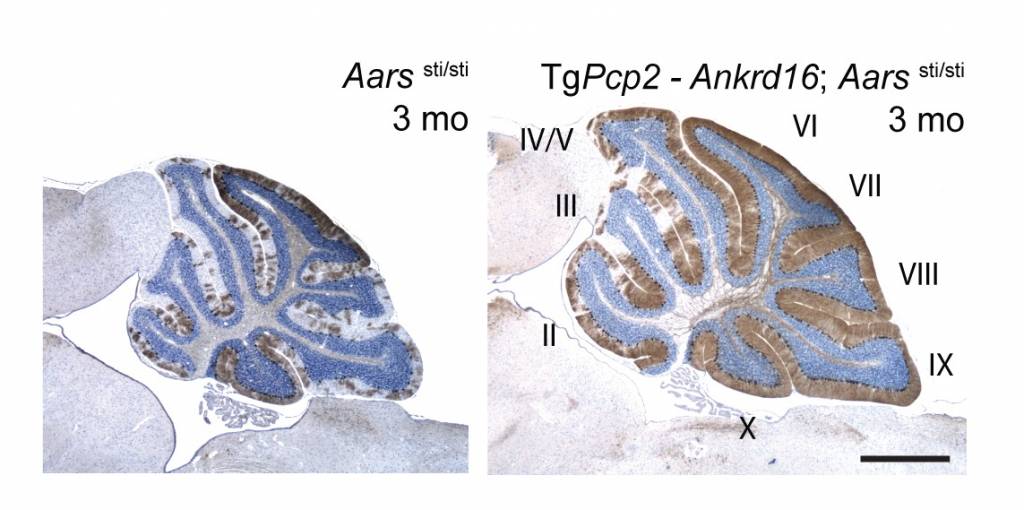
Purkinje cell loss in mice homozygous for the spontaneous sticky (sti) mutation is associated with accumulation of ubiquitinated proteins in the cytoplasm, endoplasmic reticulum, and nucleolus. We have determined that the sti molecular defect is a point mutation in the editing domain of alanyl tRNA synthetase (AlaRS). The aminoacyl tRNA synthetases establish the genetic code that links each amino acid to its cognate tRNA which bears the anticodon triplet of the code. The high accuracy of protein synthesis is largely attributable to the precision of these aminoacylation reactions, and much of this accuracy resides in the editing domains of these synthetases that clear misactivated amino acids or mischarged tRNAs. We demonstrated that the sti mutation causes an increase in mischarged serine-tRNAAla. This likely leads to random misincorporation of serine at alanine codons, ultimately causing production of heterogeneous, unfolded proteins. The loss of translational fidelity in sti mutant mice is an exciting new mechanism underlying neurodegeneration.
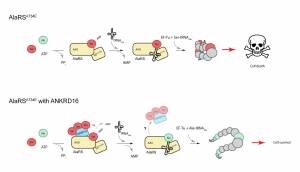
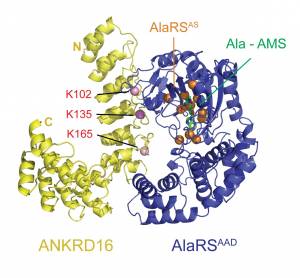
Like the mutation of Gtpbp2, Purkinje cell degeneration associated with the sti mutation varies with genetic background. Using this variation and positional cloning, we have identified the ankyrin repeat protein, ANKRD16, as a genetic modifier of sti that suppresses protein inclusion formation and neurodegeneration in mutant Purkinje cells in a gene dosage–dependent manner. Our experiments demonstrate that ANKRD16 directly interacts with AlaRS and misactivated serine is transferred to lysines on ANKRD16, thereby preventing misacylation of tRNAAla and the production of unfolded proteins. ANKRD16 is ubiquitously expressed but has lower levels in Purkinje cells suggesting that these neurons have a lower threshold for AlaRS-associated editing defects. Indeed, when Ankrd16 is deleted in the hippocampus in sti mutant mice these neurons develop protein aggregates and degenerate. We are now using various Cre lines to delete Ankrd16 in sti mutant mice to examine the effects of mistranslation on protein aggregate formation in different neuronal populations.
Work on these projects, as well as investigation of other genes identified in our screen, should continue to yield exciting insights into the mechanisms underlying neurodegeneration